Klippel-Feilův syndrom (KFS) — je vzácná vrozená porucha charakterizovaná abnormálním splynutím dvou nebo více krčních obratlů. To vede ke zkrácení krku, omezení pohyblivosti a různým kosterním a neurologickým poruchám. Syndrom je pojmenován podle francouzských neurologů Maurice Klippela a André Feila, kteří jej poprvé popsali v roce 1912.
Příčiny Klippel-Feil syndromu
Přesné příčiny SCF nejsou plně objasněny, ale předpokládá se, že k němu dochází v důsledku narušení embryonálního vývoje krční páteře během 3. až 8. týdne těhotenství. Mezi možné rizikové faktory patří:
- Genetické mutace: SCF může být způsobena mutacemi v genech GDF6, GDF3 a MEOX1. GDF6 a GDF3 ovlivňují embryonální vývoj kostí a gen MEOX1 kóduje homeobox protein MOX1, který reguluje separaci obratlů. Anomálie GDF6 a GDF3 se dědí autozomálně dominantním způsobem, zatímco mutace MEOX1 se dědí autozomálně recesivním způsobem.
- Environmentální faktory: Některé studie naznačují, že vystavení určitým chemikáliím nebo lékům během těhotenství může zvýšit riziko rozvoje SCF.
- Kombinace genetických a environmentálních faktorů.
Epidemiologie Klippel-Feilova syndromu
SCF se celosvětově vyskytuje přibližně u 1 ze 40 000 až 42 000 novorozenců, s mírnou převahou u žen.
Příznaky Klippel-Feil syndromu
Mezi hlavní příznaky SCF patří:
- Zkrácení krku
- Omezená pohyblivost krku
- Nízká linie vlasů v zadní části hlavy
- Vysoké postavení lopatek (Sprengelův syndrom)
- Skolióza nebo kyfóza hrudní páteře
- Poruchy sluchu nebo zraku
- Neurologické poruchy (v důsledku stlačení míchy nebo nervových kořenů)
Závažnost symptomů se může lišit od mírných až po závažné a závisí na úrovni a rozsahu srůstu obratlů. Klasická triáda SCF (krátký krk, nízká vlasová linie v zadní části hlavy a omezená pohyblivost krku) je pozorována u méně než 50 % pacientů.
SCF může být také asymptomatické.a děti, které nepodstoupí zobrazení krční páteře nebo které nemají zjevnou fyzickou deformaci, mohou dosáhnout dospělosti, aniž by znaly svůj stav.
Diagnóza Klippel-Feil syndromu
Diagnóza SCF je založena na klinických příznacích a zobrazovacích studiích:
- Rentgen krční páteře: dokáže detekovat fúzi obratlů a další kostní abnormality.
- CT nebo MRI: poskytují podrobnější informace o stavu obratlů, míchy a nervových struktur.
- Ultrazvuk: Dokáže detekovat související srdeční, gastrointestinální a renální abnormality u novorozenců.
- Laboratorní testy: Mohou být užitečné pro hodnocení souvisejících poruch, jako je kardiovaskulární nebo renální dysfunkce, zánětlivé procesy a krvácivé poruchy.
Léčba Klippel-Feilova syndromu
Léčba SCF je zaměřena na korekci symptomů a prevenci komplikací. Mezi klíčové přístupy patří:
- Sledování: Pravidelné prohlídky u ortopeda a neurologa k posouzení stavu páteře a neurologických funkcí.
- Fyzioterapie: cvičení na posílení šíjových svalů a zlepšení držení těla.
- Ortopedické pomůcky: krční límce nebo korzety pro podporu páteře a zabránění deformacím.
- Chirurgie: V těžkých případech může být nutná operace k dekompresi míchy, stabilizaci páteře nebo korekci deformit. Chirurgické přístupy zahrnují přední a zadní přístup a také možnost použití totální endoprotézy krční ploténky.
- Léčba doprovodných poruch: sluch, zrak, kardiovaskulární nebo genitourinární systém.
Prognóza a kvalita života
Prognóza SCF závisí na závažnosti onemocnění a přítomnosti doprovodných poruch. Pacienti s Klippel-Feilovým syndromem a srůstem krčních obratlů nad úrovní C3 mají obvykle závažnější příznaky. U pacientů s deformitou typu I se častěji vyskytnou axiální příznaky, zatímco u pacientů s deformitami typu II a III se může vyvinout myelopatie a radikulopatie.
Včasná diagnostika, správná léčba a podpora ze strany rodiny a zdravotníků hrají důležitou roli při zlepšování kvality života pacientů s SCF. Genetické poradenství může být užitečné pro rodiny s dědičnými formami SCF.
Diagnostika a léčba v Izraeli – proč právě na klinice Hadassah?
- Již více než 100 let jsou Hadassah a lékařská centra, která založila, lídrem v poskytování lékařských služeb v Izraeli.
- Více než 50 % výzkumu v oblasti nejnovějších lékařských technologií a léčebných metod v Izraeli provádějí lékaři Hadassah.
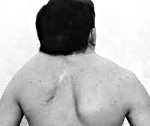
Klippel-Feilův syndrom — geneticky podmíněná anomálie ve struktuře krční páteře, včetně snížení počtu a srůstu obratlů. Klinicky se projevuje vizuálně určeným zkrácením šíje, nízkým postavením vlasové linie na zátylku a omezenými pohyby hlavy. Klippel-Feilův syndrom je zpravidla kombinován s jinými vrozenými anomáliemi skeletu a somatických orgánů. Na diagnostice se podílejí různí specialisté, včetně RTG, CT a MRI páteře, genetické analýzy a rozšířeného vyšetření vnitřních orgánů (srdce, ledviny, plíce, mozek). Konzervativní léčba se provádí pomocí masáže, cvičební terapie a fyzioterapie. Je možná chirurgická léčba – operace cervikalizace.
ICD-10
Q76.1 Klippel-Feilův syndrom
- Příčiny
- Příznaky
- diagnostika
- Léčba Klippel-Feilova syndromu
- Předpověď
- Ceny za ošetření
Přehled
Klippel-Feilův syndrom je vrozená, geneticky podmíněná patologie krční páteře, sestávající z fúze (synostózy) a snížení počtu obratlů. Nejtypičtějším a stálým příznakem syndromu je výrazné zkrácení krku, proto se v lékařské praxi označuje také jako syndrom krátkého krku. Ve většině případů se kombinuje s dalšími vývojovými anomáliemi pohybového aparátu (torticollis, skolióza, Sprengelova choroba, hypoplazie horních končetin, syndaktylie) a vrozenými vadami vnitřních orgánů (ledviny, kardiovaskulární systém, plíce). Klippel-Feilův syndrom je vzácné onemocnění. Její incidence je přibližně 1 případ na 120 tisíc novorozenců. Syndrom byl poprvé popsán v roce 1812 ve Francii neurologem Klippelem a radiologem Feilem, jejichž jména tvořila základ pro jeho jméno.
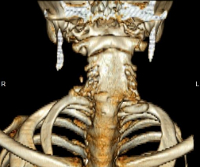
Moderní klinická neurologie klasifikuje Klippel-Feilův syndrom do 3 typů. První typ, KFS1, se vyznačuje sníženým počtem krčních obratlů. Normálně má člověk v krční páteři 7 obratlů, u KFS1 obvykle 4-5. Druhý typ – KFS2 – synostóza všech krčních obratlů, jejich splynutí s týlní kostí a horními hrudními obratli. Třetí typ, KFS3, je kombinací prvního nebo druhého se srůstem obratlů v dolní hrudní a bederní oblasti. Často jsou v cervikální oblasti pozorována další žebra a spina bifida (nestavení obratlových oblouků).
Příčiny
Klippel-Feilův syndrom je geneticky podmíněná patologie. Ojediněle se vyskytují sporadické případy syndromu. Anomálie se tvoří in utero, dokonce i v embryonálním období, v důsledku hypo- a aplazie, porušení rozdělení cervikálních segmentů a opožděné fúze v procesu tvorby obratlů. Genetické aberace jsou heterogenní. U KFS1 ovlivňují lokus q22.1 8. chromozomu, u KFS2 se nacházejí v lokusu q22.1 5. chromozomu a u KFS3 jsou umístěny v lokusu q13.31 12. chromozomu.
Nejvíce studovaným genem je GDF6, který je zodpovědný za vznik KFS1. Mutace tohoto genu vedou k narušení syntézy proteinu podílejícího se na tvorbě pohybového aparátu vytvořením hranic mezi jednotlivými kostmi. V závislosti na typu má Klippel-Feilův syndrom různý mechanismus dědičnosti: pro KFS1 a KFS3 je autozomálně dominantní, pro KFS2 je autozomálně recesivní.
Příznaky
Hlavní klinickou triádou charakterizující Klippel-Feilův syndrom je zkrácení krku, posun vlasové linie dolů na zadní straně krku a zhoršená pohyblivost páteře v krční oblasti. Závažnost zkrácení krku se může v nejtěžším případě lišit, ušní boltce dosahují k ramenům a brada dosahuje hrudní kosti, polykání a dýchání je obtížné. Vyznačuje se širokým rozkročením lopatek a často jejich zkrácením. Lze pozorovat vysoké postavení jedné z lopatek, typické pro Sprengelovu chorobu. V některých případech jsou zaznamenány anomálie svalů ramenního pletence a záhyby na krku. Ve vzácných případech se objevuje radikulární syndrom – bolest spojená s kompresí krčních míšních kořenů.
V 50-60% případů je Klippel-Feilův syndrom kombinován se skoliózou, ve 25% případů – s kostní variantou torticollis. Možná je kombinace syndromu krátkého krku s anomáliemi horních končetin (polydaktylie, syndaktylie, vrozené amputace), deformitami chodidel, defekty žeber, anomáliemi chrupu, asymetrií obličeje a dalekozrakostí. U 45 % pacientů je diagnostikována dystopie, aplazie nebo hypoplazie ledvin, hydronefróza a ektopie močovodů. U 25 % pacientů je detekována vrozená hluchota, u 20 % – rozštěp patra, u 15 % – vrozené srdeční vady (patent ductus arteriosus, VSD, ASD, dextropozice aorty). Může být pozorována aplazie nebo hypoplazie plic.
Ze strany nervového systému se může vyskytovat oligofrenie (mentální retardace), epilepsie, hydrocefalus, spina bifida, mikrocefalie, poruchy okohybnosti (strabismus, ptóza, Hornerův syndrom). Od raného věku je charakteristická svalová slabost na končetinách a synkineze – mimovolní současné pohyby obou paží, nejčastěji pouze rukou. Časem se může objevit spastická a ochablá para- a tetraparéza.
diagnostika
Ověření diagnózy se provádí na základě typické triády příznaků pozorovaných od narození, údajů z vyšetření, rodinné anamnézy a výsledků instrumentálních a genetických studií. Klippel-Feilův syndrom s podrobnou indikací stávajících doprovodných anomálií je možné stanovit pouze společnou prací mnoha odborníků: neurologa, ortopeda, genetika, kardiologa, nefrologa, pneumologa, oftalmologa.
Nejprve se provede rentgenový snímek krční páteře ve 2 projekcích. U KFS1 rentgenové snímky ve většině případů odhalí úplnou synostózu 4–5 obratlů do jednoho špatně diferencovaného konglomerátu. V některých případech jsou mezi obratli úzké světlé pruhy odpovídající nedostatečně vyvinutým ploténkám, což naznačuje částečnou synostózu, která vede k zakřivení páteře, jak dítě roste. Klippel-Feilův syndrom typu II je radiologicky charakterizován kombinací synostóz 7 krčních obratlů s asimilací atlasu a fúzí horních hrudních obratlů. K vyloučení KFS3 se provádí rentgenové snímky hrudní a bederní páteře.
CT páteře poskytuje úplnější informace o kostních anomáliích. Jeho použití v raném dětství je však omezené kvůli radiační zátěži spojené se studií. V případě potřeby lze provést MRI páteře k posouzení stavu struktur měkkých tkání postižené oblasti (kořeny, mícha). Klippel-Feilův syndrom by měl být odlišen od vrozené svalové torticollis a spinální tuberkulózy.
Diagnostický algoritmus dále zahrnuje vyšetření vnitřních orgánů: neurosonografie, MRI mozku, ultrazvuk dutiny břišní, ultrazvuk srdce, EKG, ultrazvuk nebo CT ledvin, vylučovací urografie, rentgen hrudníku. Konzultace genetika je poskytována s analýzou rodokmenu a testováním DNA.
Léčba Klippel-Feilova syndromu
Provádějí se především konzervativní léčebná opatření zaměřená na prevenci vzniku deformit páteře a zvýšení rozsahu pohybu v krku. Masáž se provádí v oblasti krční a límečku, ramenního pletence a horních končetin. Doporučuje se pravidelná pohybová terapie. Může být použita fyzioterapie. Podle indikací se provádí symptomatická léčba poruch ve fungování somatických orgánů. Pro radikulární bolest jsou předepsány analgetika a nošení límce Shantz.
Syndrom přetrvávající bolesti způsobený stlačením kořenů horními žebry je indikací k operaci. Chirurgický zákrok se provádí podle Bonola techniky a jedná se o tzv cervikalizace resekcí horních 4 žeber. Přístup je zajištěn paravertebrálním řezem probíhajícím rovnoběžně s vnitřním okrajem lopatky. Operace se provádí ve 2 fázích, zvlášť na každé straně.
Předpověď
Samotný Klippel-Feilův syndrom má příznivou vitální prognózu. Přítomnost malformací somatických orgánů výrazně komplikuje situaci a může být příčinou předčasné smrti. Funkčně je prognóza nepříznivá i přes přijatá konzervativní opatření, pacienti mají stále výrazné omezení pohybů hlavy, jehož míra závisí na typu a závažnosti syndromu. Průběh onemocnění mohou zhoršit degenerativní změny na páteři.