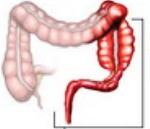
Hirschsprungova nemoc — je vrozená patologie tlustého střeva s nedostatečně vyvinutým nebo nepřítomným nervovým plexem v submukózní a svalové vrstvě celého tlustého střeva nebo jeho segmentu. Projevuje se jako chronická zácpa, nedostatek nutkání na stolici, plynatost, nelokalizované bolesti břicha, asymetrický tvar břicha a intoxikace. Diagnostikováno pomocí irrigoskopie, rektoskopie, histologie biopsie stěny tlustého střeva, anorektální manometrie. Léčí se chirurgickými metodami, pacientům se doporučuje jednostupňová nebo dvoustupňová resekce tlustého střeva s vytvořením kolorektální anastomózy.
ICD-10
Q43.1 Hirschsprungova nemoc
- Příčiny
- Patogeneze
- Klasifikace
- Příznaky Hirschsprungovy choroby
- Komplikace
- diagnostika
- Léčba Hirschsprungovy choroby
- Prognóza a prevence
- Ceny za ošetření
Přehled
Aganglionóza tlustého střeva (vrozené megakolon) je jednou z běžných anomálií ve vývoji trávicích orgánů. Podle pozorování je prevalence Hirschsprungovy choroby v populaci novorozenců od 1:30 000 do 1:2 000 U 90 % pacientů onemocnění debutuje před 10. rokem života. U chlapců je anomálie zjištěna 4,32krát častěji než u dívek. Ve 29,0-32,7 % případů je patologie spojena s jinými vývojovými vadami, u 9 % pacientů se rozvíjí aganglionóza tlustého střeva v rámci Downova syndromu. Porucha byla poprvé popsána v dílech dánského pediatra Haralda Hirschsprunga v roce 1888. Relevance včasného odhalení onemocnění je způsobena závažností jeho komplikací.
Příčiny
Hirschsprungova choroba má polyetiologický původ, stále se objasňuje role předpokládaných a produkujících faktorů přispívajících k rozvoji aganglionózy stěny tlustého střeva. S největší pravděpodobností je vrozená vada důsledkem kritického poškození genů, které regulují tvorbu nervových struktur tlustého střeva. Podle odborníků v oblasti praktické proktologie je výskyt Hirschsprungovy anomálie usnadněn:
- Zatížená dědičnost. U 20 % pacientů má onemocnění familiární charakter. Podle výsledků molekulárně genetických studií narušují dědičné mutace genů RET, GDNF, EDN3, ENDRB migraci neuroblastů z vagového neurocrestu, což způsobuje aganglionózu střevní stěny. Ve 12 % případů onemocnění jsou pozorovány chromozomální aberace, v 18 % se vada projevuje ve struktuře dědičných syndromů.
- Dysontogeneze. Nedědičné formy onemocnění jsou spojeny s vlivem intrauterinní virové infekce, vysoké radiace, mutagenních chemikálií, které narušují diferenciaci neuroblastů. Riziko vzniku anomálie se zvyšuje s porodnickou patologií a chronickými onemocněními těhotné ženy, doprovázenými tkáňovou hypoxií – gestózou, kardiopatologií (hypertenze, srdeční selhání), diabetes mellitus.
Patogeneze
Rozvoj Hirschsprungovy choroby je způsoben poruchou embryogeneze, pravděpodobně v 7-12 týdnu těhotenství, kdy se tvoří Meissnerovy (v submukózní vrstvě tlustého střeva) a Auerbachovy (ve svalové membráně střeva) nervové pleteně. V důsledku předčasného zastavení migrace neuroblastů nebo jejich nedostatečné diferenciace jsou místo typických submukózních a myenterických plexů s ganglií zastoupeny střevní neurostruktury jednotlivými nervovými vlákny a gliovými elementy.
Čím dříve je migrace neuroblastů dokončena, tím rozsáhlejší je agangliová oblast stěny tlustého střeva. Acetylcholinesteráza se hromadí ve slizniční vrstvě, což způsobuje střevní spasmus, což je patognomický znak Hirschsprungovy anomálie. V důsledku tonického spasmu a nedostatku peristaltiky se denervovaný segment stává funkční překážkou pohybu stolice. Chronické zadržování střevního obsahu vede k neustálé zácpě a výrazné expanzi horní části střeva.
Klasifikace
Klasifikace Hirschsprungovy choroby se provádí s ohledem na anatomická a klinická kritéria. Podle lokalizace aganglionické oblasti se rozlišuje nejčastější rektosigmoidální forma onemocnění, zjištěná u 70 % pacientů, rektální (až 25 % případů anomálie), mezisoučet (3 %), segmentální ( 1,5 %) a celkem (0,5 %). Podle umístění dilatovaného střeva se rozlišuje megarektum, megasigmoideum, levostranné, subtotální a celkové megakolon, megaileum. Při diagnostice Hirschsprungovy střevní aganglionózy se berou v úvahu následující klinické příznaky:
- Dětská verze nemoci. Je detekován u téměř 90 % pacientů. Charakterizován rychlým vývojem, téměř úplnou absencí nezávislé defekace a rostoucími známkami střevní obstrukce. Operace se obvykle provádí ve dvou fázích.
- Prodloužená varianta aganglionózy. Debuty mezi dětmi. Vzhledem k malé délce denervovaného segmentu se vyvíjí pomalu. K nápravě zácpy se po dlouhou dobu používají klystýry. Je možné provést buď dvoustupňovou nebo jednostupňovou operaci.
- Latentní varianta anomálie. V dospívání se objevuje zácpa a rychle se rozvíjí chronická obstrukce tlustého střeva. Ke zmírnění zácpy jsou nutné každodenní klystýry. Průchod výkalů je obvykle obnoven ve dvou fázích.
Při kompenzovaném průběhu přetrvává nezávislé vyprazdňování po mnoho let s progresí onemocnění, dochází k 3-7denní zácpě, která vyžaduje k vyřešení laxativa nebo klystýr. U pacientů se subkompenzovaným stavem trvá zácpa bez použití laxativ a klystýru déle než 7 dní. Dekompenzace onemocnění je indikována nepřítomností nezávislé defekace a nutkáním k tomu, zhutněním střevního obsahu a tvorbou fekálních kamenů a neúčinností konzervativních metod pohybu střev.
Příznaky Hirschsprungovy choroby
Rychlost vývoje klinického obrazu onemocnění závisí na prevalenci střevního poškození. Ve většině případů se první příznaky objevují ihned po narození dítěte, ale někdy je patologie asymptomatická, nástup je pozorován již v dospívání a dokonce i v dospělosti. Hlavním projevem aganglionózy tlustého střeva je chronická zácpa a nedostatek nutkání na stolici. 50 % pacientů pociťuje nadýmání a bolesti v dutině břišní, které jsou spojeny se zadržováním stolice.
Při dlouhodobém průběhu onemocnění dochází k rozvoji břišní asymetrie a rozšíření kožní žilní sítě. U některých pacientů může být pozorována aktivní střevní peristaltika. Při Hirschsprungově aganglionóze se často objevují známky anémie – bledá kůže a sliznice, časté závratě, snížená výkonnost. Mohou být pozorovány příznaky obecné intoxikace těla: nevolnost a zvracení, bolesti hlavy. Patologie je někdy kombinována s vrozenou heterochromií (nestejné zbarvení duhovky).
Komplikace
V případě Hirschsprungovy aganglionózy dochází u pacientů k poruchám trávení a vstřebávání živin, což v kombinaci se sníženou chutí k jídlu vede k těžké hypotrofii, až kachexii. Často je zjištěna anémie z nedostatku železa. Děti vykazují zpoždění v růstu a fyzickém vývoji. Dlouhodobá stagnace stolice ve střevě vyvolává dysbakteriózu, zánětlivé změny na sliznici, které se mohou projevit jako paradoxní průjem.
Nejnebezpečnějšími komplikacemi Hirschsprungovy choroby jsou střevní neprůchodnost, dekubity stěny z fekálních kamenů a perforace střeva. Může se vyvinout toxický megakolon, který je charakterizován expanzí proximálního střeva a nadměrným růstem patogenní bakteriální flóry. Tento stav často vede k zánětu pobřišnice a sepsi v důsledku průniku střevních bakterií přes patologicky změněnou střevní stěnu. V tomto případě dochází k ostrým, difúzním bolestem břicha, opakovanému zvracení a febrilním horečkám.
diagnostika
Hirschsprungovu chorobu lze podezřívat za přítomnosti charakteristických fyzických příznaků (prohmatání těstovitého „nádoru“ a objevení se „jílového symptomu“ – jasně viditelné stopy stlačení tlustého střeva prsty přes přední břišní stěnu). Diagnostické vyhledávání zahrnuje provedení komplexního laboratorního a instrumentálního vyšetření pacienta, které umožňuje ověření diagnózy. Nejinformativnější v diagnostice aganglionózy jsou:
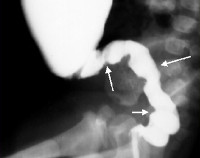
- Retrográdní radiografie tlustého střeva. Doplnění tlustého střeva rentgenovým kontrastem umožňuje vizualizaci jasného přechodu mezi dilatovanou proximální částí střeva a zúženou distální částí, která nemá inervaci. Irrigoskopie také odhalí absenci haustrace tlustého střeva.
- Endoskopické vyšetření rekta a sigmoidálního tračníku. Rektosigmoidoskopie a rektosigmoidoskopie se provádí bez speciální přípravy pacienta. U Hirschsprungovy patologie je detekována křečovitá střevní stěna a nepřítomnost stolice. Proximálně se nachází dilatovaný úsek tlustého střeva naplněný tvrdými výkaly.
- Cytomorfologická analýza podle Swansona. Histologie biopsií konečníku a tlustého střeva je „zlatým standardem“ v diagnostice onemocnění. Pro získání spolehlivých výsledků se biologický materiál odebírá ze zadní stěny střeva na vzdálenost 6 cm, počínaje od dentální linie. Kromě toho se provádí hodnocení aktivity AChE.
- Manometrická studie. Hirschsprungova aganglionóza je charakterizována absencí reflexního otevření svěrače rekta v reakci na zvýšený tlak. Je zaznamenána diskoordinace kontrakce svěračů a konečníku. Anorektální manometrie je důležitým diagnostickým kritériem a má citlivost asi 85 %.
Změny v klinickém krevním testu (leukocytóza, zvýšená ESR, toxická granularita neutrofilů) se vyskytují v případě komplikované verze onemocnění. Biochemický krevní test odhalí hypoalbuminémii a dysproteinémii. Pokud má pacient paradoxní průjem, provádí se bakteriologické vyšetření stolice k izolaci patogenních agens.
Hirschsprungovu aganglionózu je třeba především odlišit od idiopatického megakolonu. Hlavním diagnostickým kritériem je absence submukózních a intermuskulárních nervových ganglií ve střevních biopsiích. Pozornost je věnována anamnéze (počátek onemocnění v raném dětství), údajům ze studia aktivity AChE a anorektální manometrii. Pokud je to indikováno, do konzultace s pacienty se kromě proktologa zapojuje i specialista na infekční onemocnění a chirurg.
Léčba Hirschsprungovy choroby
Pacientům s potvrzenou diagnózou se doporučuje podstoupit operaci zaměřenou na obnovení střevní průchodnosti odstraněním denervované oblasti. Konzervativní metody (náprava metabolických poruch, odstranění zácpy pomocí očistných, hypertonických a sifonových klystýrů) se používají ve fázi diagnózy a předoperační přípravy. Pokud je konzervativní terapie opožděna, megakolon postupuje, stav pacienta se zhoršuje a zvyšuje se riziko pooperačních komplikací.
Při volbě objemu a techniky chirurgické intervence se bere v úvahu délka agangliového segmentu, stupeň prestenotické expanze a věk pacienta. Při operaci břicha je resekována denervovaná oblast a patologicky změněná dilatovaná část střeva a vzniká kolorektální anastomóza. S ohledem na zvolenou techniku chirurgické léčby Hirschsprungovy patologie jsou možné dva přístupy k provádění plánovaných intervencí:
- Jednostupňový provoz. Indikováno pro kompenzovanou formu onemocnění a krátkou délku agangliového segmentu. Postižené střevo se odstraní a okamžitě se vytvoří anastomóza. Výhodou jednostupňového přístupu je menší traumatizace, nicméně při nesprávném posouzení klinické situace se zvyšuje pravděpodobnost komplikací v pooperačním období.
- Dvoustupňový provoz. Doporučeno pro pacienty se subkompenzovanými a dekompenzovanými variantami onemocnění, významnými změnami v tlustém střevě nad denervovanou oblastí a rozšířeným agangliovým segmentem. V první fázi, po resekci střeva, se vytvoří kolostomie, která se po nějaké době sešije s vytvořením anastomózy tlustého střeva při rekonstrukční intervenci.
Urgentní nebo urgentní operace se provádí v případě akutní střevní neprůchodnosti, perforace střeva nebo dekubitu střevní stěny způsobeného fekálním kamenem. Intervence se provádí ve výši resekce sigmoidálního tračníku, levostranné hemikolektomie, proktektomie s uložením kolostomie nebo ileostomie. Následně je pomocí chirurgických metod obnoven průchod střevního obsahu. Ambulantní klinické vyšetření operovaných pacientů se provádí týdně po dobu jednoho měsíce po operaci, čtvrtletně po dobu jednoho roku a ročně po dobu 3 let.
Prognóza a prevence
Výsledek Hirschsprungovy choroby závisí na době jejího záchytu a stupni poškození střevních nervových ganglií. Prognóza je v případě včasné diagnózy a chirurgické korekce poměrně příznivá. Bez léčby dosahuje kojenecká úmrtnost v prvních měsících života 80 %. Vzhledem k vrozené povaze patologie nebyla vyvinuta specifická preventivní opatření. Když se objeví první příznaky onemocnění, musíte se okamžitě poradit s lékařem, abyste se vyhnuli rozvoji závažných komplikací.
Literatura
1. Klinická operační proktologie / Fedorov V.D., Vorobyov G.I., Rivkin V.L. — 1994.
2. Klinické pokyny pro diagnostiku a léčbu dospělých pacientů s Hirschsprungovou chorobou / Asociace proktologů Ruska – 2013.
3. Léčba Hirschsprungovy choroby u dospělých / Moskvitin I.S., Zaigraev B.V., Plekhanov A.N., Tovarshinov A.I. // Bulletin VSRC SB RAMS — 2012 — č. 4
4. K problematice diagnostiky Hirschsprungovy choroby / Shakumidova A.G., Mashkov A.E., Shcherbina V.I., Tsuman V.G., Semilov E.A., Sinenkova N.V. // Pediatrie. Časopis pojmenovaný po G.N. Speransky – 2006